


Nouveau règlement : certains DM de classe IIb et III sur la sellette (Partie 2)

Après avoir évoqué, dans une première partie, les grands changements apportés par le nouveau règlement DM, le cabinet de conseil Strategiqual nous présente plus en détail les procédures d'évaluation de la conformité ainsi que la notion de normes harmonisées et de spécifications communes.
Aymeric Lebon et Céline Colineaux, Strategiqual

Céline Colineaux

Aymeric Lebon
Avant la mise sur le marché d'un DM, sa conformité doit être évaluée par un Organisme Notifié (ON). L’art. 42(1) du nouveau règlement DM (RDM) introduit les procédures d’évaluation de la conformité des dispositifs qui sont définies aux annexes VIII à XI. Ces procédures connaissent quelques évolutions. En particulier :
- L’annexe II du RDM, procédure d’évaluation des dispositifs de classe I (sauf exceptions), relative à la documentation technique, détaille la partie surveillance après commercialisation.
- L’ON interviendra dans l’évaluation de la conformité des instruments chirurgicaux réutilisables mis sur le marché à l'état stérile, l’intervention se limitant aux aspects liés à la réutilisation du dispositif.
- L’option d’une évaluation de la conformité selon l’annexe VI de la directive actuelle (contrôle final) disparaît dans le RDM pour les dispositifs de classe IIa et IIb.
- Une procédure d’évaluation de la conformité spécifique aux dispositifs sur mesure a été introduite dans le RDM (annexe XI).
A noter que certains DM seront désormais soumis à des procédures spécifiques d’évaluation de la conformité, notamment :
- les dispositifs incorporant comme partie intégrante une substance qui, utilisée séparément, serait considérée comme un médicament,
- les dispositifs incorporant comme partie intégrante des tissus ou cellules d’origine humaine ou leurs dérivés,
- les dispositifs qui sont composés de substances ou de combinaisons de substances, lesquelles sont destinées à être introduites dans le corps humain par un orifice du corps ou par application sur la peau et qui sont absorbées par le corps humain ou dispersées localement.
En ce qui concerne l’évaluation de conformité, notons que le certificat d’attestation CE et le certificat CE de type, délivrés par les ON, deviennent respectivement certificat d’évaluation EU de la documentation technique et certificat d’examen UE de type (annexe XII). La déclaration CE de conformité fait désormais place quant à elle à la déclaration de conformité UE dans le RDM (annexe III).
Les ON davantage considérés comme des autorités de tutelle
Penchons-nous à présent sur les principaux changements au niveau de l’évaluation de la conformité par l’ON :
- L’examen des preuves cliniques par l’ON est accentué, avec une vérification des preuves cliniques et un lien avec les procédures spéciales pour les dispositifs concernés (chap.II, point 5.3 bis de l’annexe VIII).
- La fréquence des audits inopinés réalisés sur place par les ON chez les opérateurs économiques est établie à au moins une fois tous les cinq ans (chap.I, point 4.4 de l’annexe VIII).
- Le rôle et les responsabilités de l’ON sont de plus en plus détaillés, notamment dans le cadre de l’évaluation de surveillance intégrant l’essai des pièces et/ou des matériaux approuvés qui sont essentiels pour l'intégrité du dispositif (chap.I, point 4.5 de l’annexe VIII).
- La création d’un groupe d’experts, groupe de coordination en matière de dispositifs médicaux (GCDM), avec des tâches définies à l’art. 80, contribuera à fournir des conseils quant à la sélection du ou des échantillons représentatifs dont l’ON aura besoin dans le cadre des audits des opérateurs économiques (chap.I, point 3.3. c) de l’annexe VIII).
- Un document complémentaire au certificat d'évaluation UE fait son apparition dans le RDM en cas de modifications du dispositif si ces modifications sont susceptibles de remettre en cause la sécurité et/ou les performances du dispositif ou les conditions prescrites pour l'utilisation du dispositif (chap.II, point 5.5 bis de l’annexe VIII).
- Des dispositions administratives telles que la durée de conservation de documents qualité sont détaillées dans le RDM (chap.III, point 8 de l’annexe VIII). Ainsi, les données du dossier technique doivent être majoritairement disponibles pendant 10 ans après l’introduction sur le marché du dernier dispositif médical.
- La formalisation de procédures spéciales est détaillée dans le RDM (chap.III, point 6 de l’annexe VIII).
Normes harmonisées et spécifications communes
L’art. 4(2) fonde la conformité d’un DM aux prescriptions générales de sécurité et de performance de l’annexe I avec une référence explicite :
- aux normes harmonisées dont les références seront publiées au Journal officiel de l'Union européenne (art. 6(1))
- et, le cas échéant, aux Spécifications Communes (SC) couvertes par l’art. 7 lorsqu'il n'existe pas de normes harmonisées, que les normes harmonisées applicables ne suffisent pas ou qu'il y a lieu de répondre à des préoccupations de santé publique. On entend par SC (art. 2) : « tout document autre qu'une norme qui énonce des prescriptions techniques et/ou cliniques offrant un moyen de se conformer aux obligations légales applicables à un dispositif, à un procédé ou à un système ».
Conformément à l’art. 7, les SC peuvent être adoptées par la Commission, après consultation du GCDM, en ce qui concerne les prescriptions générales en matière de sécurité et de performances énoncées à l'annexe I, la documentation technique prévue à l'annexe II, l'évaluation clinique et le suivi clinique après commercialisation prévus à l'annexe XIII ou les prescriptions relatives aux investigations cliniques énoncées à l'annexe XIV.
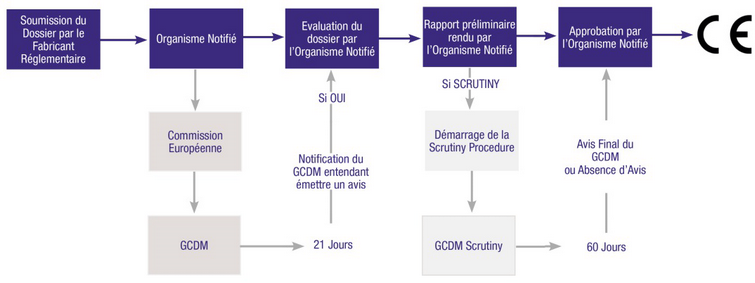
Certains DM de classes IIb et III font l’objet de procédures d’évaluation spéciales, avec émission d’un avis scientifique de la part du GCDM.
En ce qui concerne les procédures spéciales, les évolutions suivantes sont à noter : les articles 43 bis et 44 introduisent la procédure de consultation dans le cadre de l'évaluation clinique pour certains dispositifs des classes IIb et III. Cela concerne notamment les dispositifs implantables de classe III avec des exceptions à la clef, telles que les demandes de renouvellement de certificat, la modification d’un dispositif médical existant sans impact sur la balance bénéfice/risque ou l'existence de spécifications communes.
L’infographie reproduite ci-dessus présente cette procédure détaillée dans l’annexe VIII (point 6.0.) dont l’objectif du groupe d’experts (GCDM) est d’émettre un avis scientifique à propos de la détermination du rapport bénéfice/risque, de la cohérence avec la ou les indications médicales et du plan de suivi clinique après commercialisation (SCAC).




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





