


Renouvellement du marquage CE des DM et nouveau règlement européen

De nombreux fabricants s'interrogent à l'approche de mai 2020, quant au renouvellement du marquage CE de leur dispositif médical. Le niveau d'exigence des organismes notifiés paraît parfois injustifié. Fort de son expérience, Gérard Sorba nous fait part de ses recommandations.
Par le Dr Gérard Sorba, Président du Groupe MultiHealth
Les informations qui sont livrées aux industriels du dispositif médical (DM) sur l’entrée en application du nouveau règlement européen (MDR) sont parfois contradictoires. En pratique, pour les nouveaux dispositifs médicaux, le MDR n’est applicable qu’à partir de mai 2020. Ces DM devront respecter obligatoirement les nouveaux textes et être évalués par des organismes notifiés (ON) redésignés par les autorités en vertu du MDR. Pour l’instant, seuls six ON ont été audités et ont obtenu leur redésignation. Selon les dernières estimations, seulement une vingtaine d’organismes notifiés devraient satisfaire aux nouvelles exigences européennes.
Les questions posées concernent surtout le renouvellement des marquages CE qui arrivent à échéance entre aujourd’hui et le 26 mai 2020.
Étant donné que le MDR a été publié depuis plusieurs mois et que « nul n’est censé ignorer la loi », les fabricants doivent anticiper l’entrée en application des textes et se mettre en conformité dans le laps de temps qui la précède.
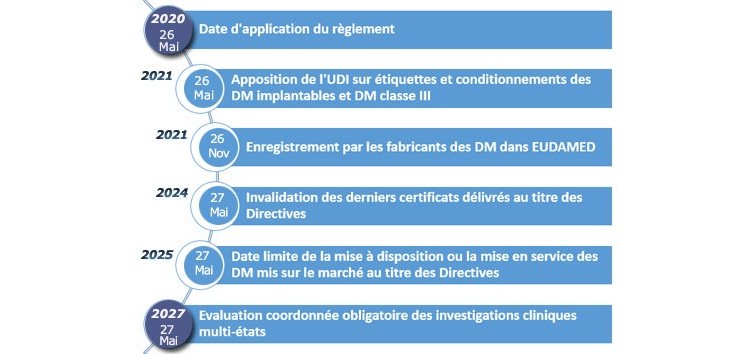
Echéances à respecter au delà de la date d'application du règlement (EU) 2017/745 (source ANSM).
Dans le cadre du renouvellement du marquage CE d’un dispositif médical, les organismes notifiés ont tendance à demander aux fabricants un maximum de données cliniques et à exiger une évaluation dont le niveau de preuve est maximal. Ici, le « Gold Standard » est l’étude clinique comparative versus placebo ou produit de référence. Or, très souvent, cette démarche est inutile et les fabricants de DM se trouvent démunis face à la menace implicite de non-renouvellement de leur marquage CE.
La véritable problématique se trouve ailleurs...
Il se trouve que les organismes notifiés n’ont pas les compétences en interne pour juger du type de méthodologie clinique nécessaire pour évaluer ou réévaluer un DM déjà sur le marché. Ils font appel à des « experts extérieurs » pour savoir quel design d’étude demander au fabricant. Ces experts médicaux extérieurs sont souvent des spécialistes de l’expertise clinique, mais quasiment jamais des experts méthodologiques. De ce fait, ils préconisent presque systématiquement, par défaut, des études cliniques comparatives en double aveugle « Gold Standard ». Le fabricant se voit dans la nécessité, soit d’appliquer à la lettre les préconisations, soit de discuter les attentes de l’organisme notifié qui aura du mal à admettre son incompétence dans ce domaine.
Nous recommandons au fabricant la démarche suivante :
- demander au contact de l’organisme notifié la justification de la demande, d’un point de vue méthodologique ;
- en cas de refus : demander le nom de l’expert afin de pouvoir entrer en contact avec lui ;
- faire appel à un expert clinique et à un expert méthodologique afin de proposer une solution acceptable pour l’organisme notifié, sous peine d’arriver dans une impasse.
Il faut préciser que la demande d'un organisme notifié peut tout à fait être rejetée par un comité de protection des personnes (CPP).
Citons ici le cas concret d'une étude clinique comparative versus placebo qui avait été demandée par un organisme notifié, et dont le protocole, pourtant validé par ce dernier, a été refusé par le CPP. Les motifs invoqués étaient le risque pour les patients et l'absence d'intérêt d'une telle évaluation pour le DM. Au final, nous avons proposé à l’organisme notifié de réaliser d’une part une étude observationnelle dont la méthodologie se rapproche de celle d’une investigation clinique mais qui permet au DM d’être évalué dans des conditions réelles de prescription, et d’autre part un suivi de la matériovigilance conformément au nouveau règlement européen. Cette proposition a été acceptée par l’expert mandaté par l’organisme notifié et le fabricant va pouvoir obtenir son renouvellement de marquage CE. Le seul problème est que l’industriel a perdu 6 mois dans ce dossier.
Quelle méthodologie pour un suivi clinique après commercialisation ?
Le nouveau règlement européen laisse le choix aux fabricants. L’objectif est de pouvoir démontrer un suivi afin d’améliorer la documentation et donc l’information du public et de modifier éventuellement la notice d’utilisation du DM.
Le « Gold Standard » de suivi clinique d’un dispositif médical n’est pas obligatoirement l’étude comparative. Il est donc fondamental, en fonction des caractéristiques du DM et de son utilisation chez le patient, de définir la meilleure méthodologie, cette dernière pouvant très bien être observationnelle. L’avis d’un expert clinicien et d’un expert méthodologique clinique et statistique est fondamental pour faire économiser du temps et de l’argent au fabricant.




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





