


RDM : comment gérer les évènements de sécurité dans le cadre d’une IC ?

Le RDM définit les modalités d'enregistrement et de notification des évènements indésirables qui surviennent lors d'une investigation clinique (IC). L'AFCROs rappelle ici comment ils se répartissent et prodigue plusieurs conseils aux promoteurs pour leur permettre de satisfaire à leurs obligations.
Par Carla Lippens du groupe AFCROs-DM

Carla Lippens (crédit photo : AFCROs).
Depuis son entrée en application, le RDM 2017/745 a notamment permis de renforcer les règles de vigilance afin de garantir la sécurité des dispositifs médicaux dans l’Union européenne (UE). Le processus de gestion des évènements indésirables (EI) survenant lors d’investigations cliniques (IC) fait, entre autres, partie de ces mesures. Régi par l’article 80 du règlement, ce processus comprend une identification stricte des évènements à enregistrer tout au long de l’investigation ainsi que des évènements de sécurité à notifier aux autorités compétentes (AC), et en définit les modalités de notification suivant les cas d’investigation : IC portant sur un DM non marqué CE ou marqué CE mais utilisé hors destination, IC réalisée dans le cadre du suivi clinique après commercialisation (SCAC) du DM, IC comprenant ou non des procédures additionnelles invasives ou lourdes et IC également en cours dans un pays tiers non européen.
Au vu de ces modalités d’enregistrement et de notification, il est donc important pour les promoteurs de :
- connaître et comprendre les exigences règlementaires associées et identifier les dispositions à suivre pour la notification des évènements reportables aux autorités compétentes ;
- bien préparer les documents d’IC mis à la disposition des investigateurs qui permettront d’enregistrer les informations relatives aux évènements de sécurité pouvant survenir durant l’étude ;
- présenter aux investigateurs une marche à suivre claire afin d’assurer une transmission efficace des informations dans les délais exigés.
Focus sur le MDCG 2020-10
Publié initialement en mai 2020 et revu en octobre 2022, le MDCG 2020-10/1 (Guide 2020-10/1 du Groupe de coordination en matière de dispositifs médicaux) vise à clarifier et à harmoniser entre les états membres les exigences relatives aux modalités de déclaration aux AC des évènements de sécurité survenant dans le cadre d’investigations cliniques. Il concerne les IC visant à établir la conformité des DM, que ce soit en pré-marché ou dans le cadre du SCAC, ainsi que les IC ayant débuté conformément aux Directives DM (93/42/EEC) et DMIA (90/385/EEC) avant le 26 mai 2021 et le cas particulier des DM utilisés dans le cadre d’essais cliniques médicaments.
La révision du guide MDCG 2020-10 publiée en octobre 2022 a permis de renforcer les points suivants :
- Définition de certains évènements spécifiques dont les "incidents" et "incidents graves" et introduction de la notion de "nouveaux résultats" ainsi que les modalités de déclaration associées,
- Clarification des modalités de déclaration d’un EIG (EI grave) survenant dans le cadre d’une IC SCAC en fonction du lien de causalité avec la procédure d’investigation qui le précède,
- Précision sur le fait d’évaluer le lien de causalité d’un EI avec le DM à l’étude ou la procédure d’investigation de façon indépendante,
- Apport d’informations sur l’identification du statut d’un évènement à renseigner par le promoteur lors de la notification aux AC,
- Identification des codes IMDRF (International Medical Device Regulators Forum) applicables pour la notification des "signes/symptômes cliniques", "impact clinique" et "problème lié au dispositif".
Ce guide s’accompagne, par ailleurs, d’un formulaire de rapport d’évènement (MDCG 2020-10/2), devant être utilisé par les promoteurs lors des déclarations d’évènements de sécurité rapportés en accord avec l’article 80 du RDM, et ce jusqu’à ce que la plateforme EUDAMED soit fonctionnelle. La transmission de ce formulaire à toute autorité nationale compétente des états membres de l’UE où l’IC est réalisée, est requise, ainsi que la diffusion aux AC suisse et turque si l’IC est conduite dans ces pays.
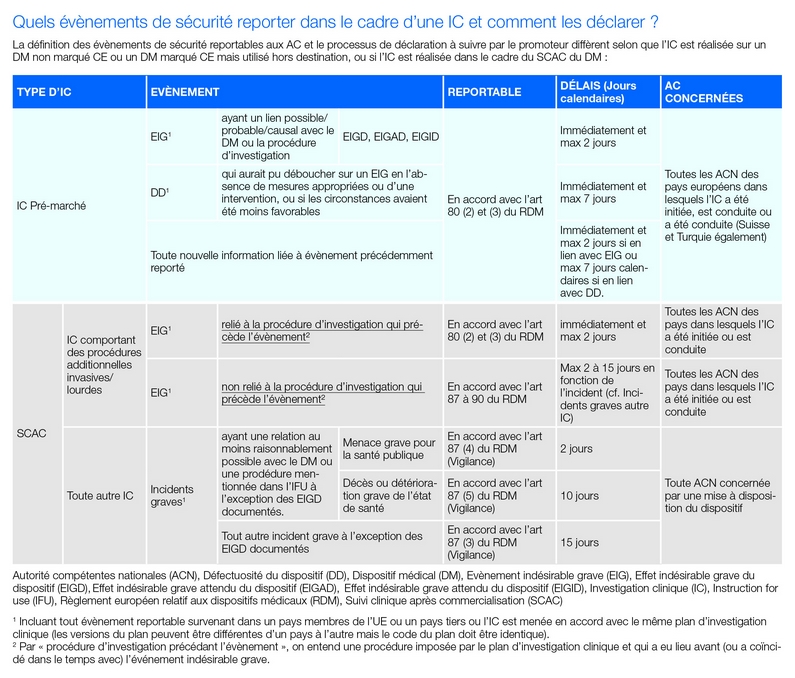
Préparer les investigateurs à la procédure de gestion des EI
Afin de garantir la transmission efficace des informations nécessaires à l’élaboration du rapport de déclaration d’évènement aux AC (MDCG 2020-10/2), le promoteur doit, en premier lieu, s’assurer que les documents mis à la disposition des investigateurs sont conçus et rédigés de manière à collecter tous les détails pertinents d’un évènement, selon les mêmes termes et formats, et avec les mêmes propositions de choix, que ceux employés dans le formulaire de déclaration aux AC. Ces dispositions concernent principalement les formulaires d’enregistrement des EI et des défectuosités d’un dispositif (DD), ainsi que le formulaire de rapport des EIG, lequel peut également être utilisé pour les DD qui auraient pu déboucher sur un EIG en l’absence de mesures appropriées ou d’une intervention, ou dans des circonstances moins favorables.
Ainsi par exemple :
- Toutes les dates doivent être collectées au format JJ/MM/AAAA,
- Le genre du patient peut être défini comme Femme/Homme/Autre/Inconnu,
- Le lien de causalité d'un évènement avec le DM ou la procédure d'investigation est à définir comme non relié, possible, probable ou causal au regard des définitions strictes énoncées dans le MDCG 2020-10/1
- Le bras d'investigation dans lequel l'évènement est survenu peut être Groupe Test, Comparateur, Aveugle ou Non applicable,
- Le statut d'actualisation d'une information peut être Ajouté (A=Added) ; Supprimé (D=Deleted) ; Modifié (M=Modified) ou Inchangé (U=Unchanged) ;
- Le statut d'un évènement est à définir comme résolu avec séquelles, en cours, décès.
L’utilisation d’une terminologie standardisée étant fortement encouragée lors des déclarations aux AC sur la base des codes IMDRF pertinents, il peut être aussi envisagé par le promoteur d’intégrer cette terminologie dès le rapport de notification d’un évènement complété par l’investigateur, du moins concernant les « signes/symptômes cliniques » et « l’impact clinique ».
Enfin, la formation des investigateurs au processus de gestion des évènements de sécurité dans le cadre d’une IC reste une étape essentielle. Une bonne compréhension par les investigateurs des évènements à enregistrer/reporter, des moyens mis à leur disposition (e-CRF, formulaire, procédures…), des délais requis, et des personnes à informer, permet en effet d’optimiser la mise en application de ce processus et d’en assurer l’efficacité.
En conclusion…
Partie intégrante de la vigilance, la gestion des évènements de sécurité lors de la conduite d’investigations cliniques nécessite la mise en place, par le promoteur, d'un processus efficace en accord avec les exigences applicables du RDM selon le type d’étude. Une définition précise de ce processus dès la conception des documents d’IC et une bonne compréhension de celui-ci par les investigateurs favoriseront une mise en application réussie.
Sources :
https://health.ec.europa.eu/system/files/2022-11/md_mdcg_2020-10-1_guidance_safety_reporting_en.pdf
https://ansm.sante.fr/vos-demarches/industriel/dispositifs-medicaux-vigilance-des-investigations-cliniques




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





