


Revue de détail des nouveautés de la future réglementation

La future réglementation européenne, dont l'entrée en vigueur a encore été repoussée, suscite de nombreux événements d'information ce trimestre, tous destinés à aider les fabricants à comprendre ce qui les attend. C’était le cas de la journée de conférences organisée à Paris le 25 octobre par DM Experts.
Avec la nouvelle réglementation européenne sur les dispositifs médicaux (RDM) et les dispositifs de diagnostic in vitro (RDIV), il est vital pour les industriels du secteur d’anticiper les actions qu’ils vont devoir mener dans les prochaines années. Mais pour cela, il faut d’abord connaitre les textes et surtout en comprendre les implications.
C’est pourquoi DM Experts, réseau de consultants spécialisés dans les DM, a décidé d’organiser une journée de conférences, dédiées aux principaux aspects des futurs règlements.
Une entrée en vigueur repoussée à l’été 2017
Première information : Richard Van den Broeck, directeur de la fédération belge des technologies médicales (beMedTech), a révélé avoir appris, la veille de son intervention, que l’entrée en vigueur de la nouvelle réglementation serait très probablement repoussée juste avant l’été 2017… au mieux. La date d’application serait donc repoussée d’autant, c’est-à-dire à la mi-2020 pour le RDM et la mi-2022 pour le RDIV (les périodes de transition étant respectivement de 3 et 5 ans).
Au fil des années, les textes pourront subir des modifications de la part de la Commission au travers d’actes "délégués" et "d’exécution", selon respectivement l’apparition d’informations nouvelles et le besoin de détailler certains aspects, en vue d’une mise en œuvre correcte des règlements. M. Van den Broeck précise que l’association MedTech Europe souhaite encore éclaircir plusieurs points concernant notamment le timing, l’UDI (identification unique), et l’évaluation clinique, particulièrement problématique pour les DM DIV. Le RDIV suscite d’ailleurs encore de nombreuses discussions.
Concernant la mise en œuvre des règlements dans les états membres, M. Van den Broeck prédit des difficultés surtout en matière d’adaptation des cadres légaux nationaux, de traçabilité, de matériovigilance, et d’organismes notifiés (ON).
Désignation des ON : un processus de plus de 2 ans
A propos d’organismes notifiés, Sophie Tabutin (BSI France) a expliqué le nouveau processus de désignation des ON, qui devrait prendre au moins deux ans à compter de l’entrée en vigueur. Les fabricants ne pourront donc pas obtenir de certificats sous le nouveau règlement avant 2019, voire 2020. Quant aux certificats émis pendant la période de transition de 3 ans, sous les directives actuelles, ils auront une durée de validité maximale de 4 ans après la date d’application (soit 2024).
Le timing est donc serré, et Mme Tabutin invite les fabricants à préparer leur transition sans tarder. La première chose à faire est de vérifier que son ON a bien l’intention d’être désigné sous le RDM, et pour quels types/classes de DM. Il faut surtout bien réfléchir à la stratégie à adopter pour les produits en cours de développement, selon la date d’achèvement de celui-ci.
De nouvelles règles de classification
Isabelle Douchet (consultante) a passé en revue les changements relatifs à la classification et aux procédures d’évaluation de la conformité. On retiendra d’abord qu’une classification peut ne pas être définitive. A la demande d’une Autorité Compétente, la commission peut en effet effectuer des reclassifications au travers du panel d’experts GCDM (Groupe de Coordination en matière de DM), qui est investi de diverses missions, dont celle de surveiller les ON.
On note de nouvelles règles de classification, une montée en niveau de classe pour certains types de DM (logiciels notamment), et l’intégration de nouveaux types de produit (solutions de conservation d’organes ou d’embryons, dispositifs destinés à éclairer le corps humain, nanomatériaux et dispositifs invasifs pour inhalation). Concernant l’évaluation de la conformité, les annexes ont été simplifiées avec un système qualité largement décrit.
Ces changements vont obliger les fabricants à revoir toutes les procédures d’évaluation.
Anne-Laure Bailly (TechMD.fr) a expliqué les nouvelles exigences en matière de données cliniques. Elle a évoqué la procédure "Scrutiny", d’évaluation clinique renforcée des DM à haut risque (sous pilotage de groupes d’experts émanant du GCDM), ainsi que le nouvel outil que représentent les "Spécifications communes". Celles-ci ont pour objet de compléter les normes harmonisées absentes ou insuffisantes, avec un caractère technique, mais aussi clinique. Enfin, les DM de classe III et implantables exigeront la rédaction d’un RCSPC (Résumé des Caractéristiques de Sécurité et des Performances Cliniques), qui sera globalement un résumé de l’évaluation clinique, publié sur la base Eudamed.
Une évaluation clinique désormais continue
Mais le gros changement de fond, c’est le fait que l’évaluation clinique va devenir "continue" (jusqu’à l’après-commercialisation), avec la notion de cycle de vie (comme pour les médicaments), des mises à jour régulières, et des liens avec la gestion des risques et celle de la qualité. Par ailleurs, la notion d’équivalence devient beaucoup plus marginale.
Pour les investigations cliniques, il faut retenir que l'obtention du marquage CE nécessitera davantage d’études, avec quelques nouveautés comme la nécessité d’une assurance pour le responsable promoteur, ou encore le procédé de demande unique via un système électronique centralisé par lequel passeront les événements indésirables graves des DM.
Un guide MedDev 2.7/1 Rev. 4 pour bien anticiper
La bonne nouvelle, c’est que le contenu du guide MedDev 2.7/1 Rev. 4 (paru cet été) anticipe le futur règlement, en fournissant une méthodologie d’évaluation clinique dynamique, tout au long du cycle de vie du DM, intégrée dans la gestion de la qualité.
Pour espérer avoir les résultats des investigations cliniques au moment de l’application du règlement, les fabricants ont tout intérêt à revoir dès maintenant leurs évaluations cliniques à la lumière de ce guide.
De responsabilités accrues pour les mandataires
Aurélien Bignon (BioM Advice) a présenté les nouvelles responsabilités des opérateurs économiques. Les obligations se multiplient pour les fabricants, concernant leur couverture financière (assurance), les personnes responsables de la conformité (voir cet article), l’enregistrement d'informations dans la base Eudamed (UDI), le choix d’un mandataire unique pour les DM de même groupe générique, etc. Mais le futur règlement fait aussi état de nouvelles obligations pour les mandataires, les distributeurs, les importateurs et les assembleurs/stérilisateurs de kits.
Le mandataire, en particulier, n’est plus seulement un point de contact des autorités. Il est investi de très nombreuses responsabilités. On peut d’ailleurs s’attendre à ce que les tarifs des mandats augmentent, voire que certains mandataires disparaissent.
Une révolution pour le diagnostic in vitro
Enfin, Muriel Gonidec (consultante), a relevé les changements, très nombreux, du RDIV par rapport à la directive actuelle (98/79/CE). Une véritable révolution qui se traduit d’ailleurs par un plus grand nombre de points communs avec le RDM. Gros changement côté classification : les listes A et B, jugées trop rigides, laissent la place à des classes A, B, C et D, selon un niveau de risque croissant en termes de santé publique et individuelle. La refonte des définitions, qui passent de 10 à 73, aboutit notamment à l’intégration des logiciels, des tests génétiques et des dispositifs de diagnostic compagnon.
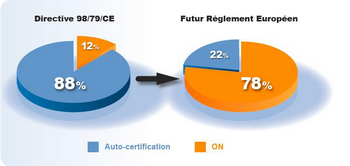
Extrait d'un communiqué publié par le SIDIV.
Seuls les DM DIV de classe A bénéficieront d’une auto-certification. Au final, d’après le SIDIV (Syndicat de l'industrie du diagnostic in vitro), 78 % des produits nécessiteront leur validation par un ON, contre 12 % actuellement. Un bouleversement pour les industriels du secteur ! D’autant plus qu’il va falloir maintenir la conformité dans le temps, avec des impacts sur l’ensemble du cycle de vie du produit.
En outre, l’évaluation des performances sera bien plus conséquente qu’auparavant, avec la rédaction de rapports sur la validité scientifique, les performances analytiques, et les performances cliniques (dont l’étude sera généralement obligatoire). Il faudra également publier un RCSPC pour les classes C et D. Sans parler de l’obligation pour le fabricant d’une surveillance pro-active après commercialisation.
Si la période de transition est plus longue pour le RDIV, il est tout aussi urgent pour les fabricants de se préparer, surtout pour les nouveaux produits, à cause notamment des exigences renforcées en matière d’évaluation clinique.
Denys Durand-Viel (DM Experts), Valérie Dumez (GS1) et Maxime Rondot (HIBC) ont présenté les tenants et aboutissants de l'UDI (Unique Device Identifier). On retiendra surtout qu'il est là-aussi urgent d'anticiper. La plupart des informations essentielles figurent dans cet article.
D’autres conférences à venir
DM Experts a prévu d’organiser des demi-journées de conférences pour traiter individuellement ces différents sujets et d’autres en rapport avec la future réglementation. Pour réduire les frais de participation, il devrait être possible d’assister à ces conférences à distance à partir d’un PC, de façon interactive.




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





