


Quelles différences entre l’UDI européen et son modèle américain ?

Agréée des deux côtés de l’Atlantique pour attribuer des codes UDI, l’association HIBCC a publié un document détaillant les différences entre les règlements européens et américain. On peut notamment s’interroger sur la base de données EUDAMED, censée être opérationnelle dans moins de quatre ans.
Evoqué dans l’article 24 et l’annexe V de la proposition de nouveau règlement européen sur les DM, le système d’identification unique (UDI) est calqué sur celui adopté aux Etats-Unis par la FDA. Son but principal est identique : assurer la traçabilité des dispositifs médicaux.
Il va s’agir pour les fabricants, d’une part de coder leurs produits, et d’autre part de renseigner la base de données publique EUDAMED.
[Mise à jour du 31 octobre 2016]
Suite à la publication de cet article, GS1 nous a fait parvenir le droit de réponse suivant : "GS1 est aujourd’hui le principal standard d'identification des dispositifs médicaux en France, avec des applications spécifiquement adaptées aux besoins métiers des professionnels de santé et conformes aux pré-requis des règlementations sanitaires, d’où leur reconnaissance par des législateurs du monde entier (FDA aux Etats-Unis, ANMAT en Argentine, ANVISA au Brésil, NEHTA en Australie, Ministère de la Santé en Turquie, etc…)".
L’UDI devra apparaître sous forme de code optique (Datamatrix par exemple) et de texte lisible, sur l’emballage de l’unité d’utilisation et tous les niveaux d’emballage supérieurs (sauf les cartons de transport), ainsi que sur le DM lui-même s’il est réutilisable. Chaque code UDI est composé d’une partie statique, l’identifiant "dispositif" (DI), propre à un fabricant et à un dispositif, et d’une partie dynamique, l’identifiant "production" (PI). Le format de ce code sera différent selon l’entité choisie pour l’attribuer. Pour l’instant, les fabricants ont le choix, comme aux Etats-Unis, entre les organismes GS1, HIBCC et ICCBBA. GS1 est le standard d’identification des produits de la grande distribution, le système HIBC a été développé spécifiquement pour les produits de santé et ICCBBA est utilisé pour la traçabilité du sang.
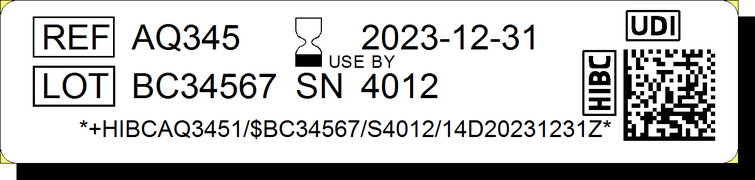 Dans le cas du standard HIBC (Health Industry Bar Code), la partie DI ("HIBAQ3451" dans l'exemple ci-contre) est composée du code d’identification du fabricant (LIC pour Labeler Identification Code), du code produit (PC) et d’un chiffre (UoM pour Unit of Measure) indiquant le niveau d’emballage. HIBC met en avant le fait que le code produit peut être composé de 1 à 18 caractères alphanumériques, qui permettent la création d’un nombre de codes quasiment illimité. De son côté, le standard GS1 utilise le code GTIN, qui offre jusqu’à 13 chiffres pour l’identification du produit.
Dans le cas du standard HIBC (Health Industry Bar Code), la partie DI ("HIBAQ3451" dans l'exemple ci-contre) est composée du code d’identification du fabricant (LIC pour Labeler Identification Code), du code produit (PC) et d’un chiffre (UoM pour Unit of Measure) indiquant le niveau d’emballage. HIBC met en avant le fait que le code produit peut être composé de 1 à 18 caractères alphanumériques, qui permettent la création d’un nombre de codes quasiment illimité. De son côté, le standard GS1 utilise le code GTIN, qui offre jusqu’à 13 chiffres pour l’identification du produit.
La partie PI, qui suit la partie DI, identifie l’unité de production d’un dispositif. Il intègre le numéro de lot ("BC34567" dans l'exemple), le numéro de série ("4012"), l'identifiant de logiciel et/ou la date de fabrication et/ou d'expiration ("20231231").
Dans le document de HIBCC qui détaille les différences entre l’UDI en Europe et l’UDI aux Etats-Unis, on note quelques différences concernant le champ PI, les règles d’étiquetage, les niveaux d’emballage, avec notamment des exemptions de marquage en cas de contraintes spatiales, que n’autorise pas le règlement américain.
Une base de données en devenir
Du côté de la base de données EUDAMED, il est prévu un fonctionnement similaire à celle de la base GUDID de la FDA, avec un accès public et gratuit, une soumission directe et automatisée des données par les fabricants, avec l’intégration du code DI et de certaines informations relatives au fabricant et au dispositif.
Contrairement à la base GUDID, il sera possible d’utiliser un DI virtuel pour les DM non labellisés, de spécifier le nombre de réutilisations possibles, ou encore d'indiquer une URL pour les instructions d’usage par exemple.
Ceci dit, il reste des zones d’ombre. La nomenclature n’est pas encore décrite ; ce qui laisse supposer que les négociations avec l’agence GMDN (organisme en charge de définir la nomenclature internationale des DM) ne sont pas terminées. Il va aussi falloir résoudre des problèmes de langues, et surtout composer avec des spécificités nationales. Harald Oehlmann, du support technique de HIBCC en Europe, est plutôt pessimiste : « Tout cela pourrait prendre plus de temps que prévu, et il est difficile d’imaginer au final un outil aussi efficace que la base GUDID. »
Si le règlement entre effectivement en vigueur cette année, son application se fera en 2019, avec l’obligation d’enregistrement des DM dans la base un an et demi après (soit entre 2020 ou 2021). Quant aux délais d’obligation de marquage, ils varient selon le type de DM : 2020, 2022 et 2024 pour les emballages respectivement des DM de classe III (et implants), de classe IIa/IIb, et de classe I. Dans tous les cas, il faut rajouter deux ans pour l’obligation de marquage direct des produits.




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





