


Conformité du SMQ d’un « legacy device » aux Règlements (UE) 2017/746 et 2024/1860

Cet article résume un mémoire de stage effectué dans le cadre du Master Ingénierie de la Santé de l'Université de Compiègne (UTC). Il décrit la démarche suivie au sein de la start-up Genotropy, pour mettre en conformité le SMQ d’un legacy device au règlement européen 2017/746 et à son amendement 2024/1860.

De g. à d. Charline Viguier, Elem Ayne et Julie Follet
Par Charline Viguier (UTC), Elem Ayne (ACR Medical) et Julie Follet (UTC)
Le guide MDCG 2022-8 définit les Dispositifs Médicaux de Diagnostic In Vitro (DMDIV) "Legacy devices" comme mis sur le marché sous la directive 98/79/CE entre le 26 mai 2022, date d’entrée en application du Règlement (UE) 2017/746 (IVDR), et la date d'expiration de la période de transition qui leur est applicable en fonction de leur classe de risque. Ils doivent être dotés d’un certificat CE médical délivré sous l’ancienne directive avant le 26 mai 2022 à la suite d’une auto-certification ou d’une évaluation par un organisme notifié. Ils doivent aussi respecter les exigences du Règlement relatives à la mise à jour du Système de Management de la Qualité (SMQ), la surveillance après commercialisation, la vigilance, la surveillance du marché, et l'enregistrement des opérateurs économiques et des dispositifs.
Pour prévenir les risques de pénurie consécutifs à une transition règlementaire plus lente que prévue, le Règlement (UE) 2024/1860, 4ème amendement impactant les dispositions transitoires fixées notamment à l'article 110.3 du Règlement (UE) 2017/746, prolonge de 2 ans la fin des périodes de transition des « legacy devices » depuis son entrée en vigueur le 9 juillet 2024, modulo le respect de nouvelles conditions (cf. Figure 1).
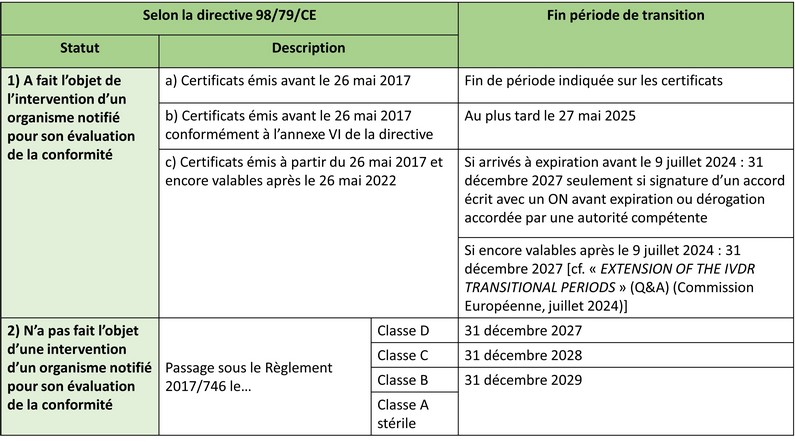
Figure 1 - Dates de fin de périodes de transition du Règlement (UE) 2024/1860 selon conditions (source UTC).
Un DMDIV qui évolue en classe D sous Règlement (UE) 2017/746
La mise en conformité aux exigences relatives à la surveillance après commercialisation, la vigilance et l’enregistrement des opérateurs économiques et des dispositifs, non impactées par l’amendement, a été menée au sein de la start-up rouennaise Genotropy qui a développé un dispositif destiné à la détermination du Rhésus D fœtal (AIO-RHD Fetal DNA Kit), marqué CE médical selon la directive 98/79/CE (annexe IV, liste A, qui évolue en classe de risque D sous le Règlement (UE) 2017/746).
La démarche suivie, objet de cet article, a débuté par la mise en conformité du système de surveillance après commercialisation de l’entreprise avec les exigences du Chapitre VII section 1 du Règlement (UE) 2017/746 (cf. Figure 2). Il s'agit de surveiller de façon proactive et réactive le rapport bénéfice-risque du dispositif tout au long de son cycle de vie pour s’assurer qu’il reste favorable au patient.
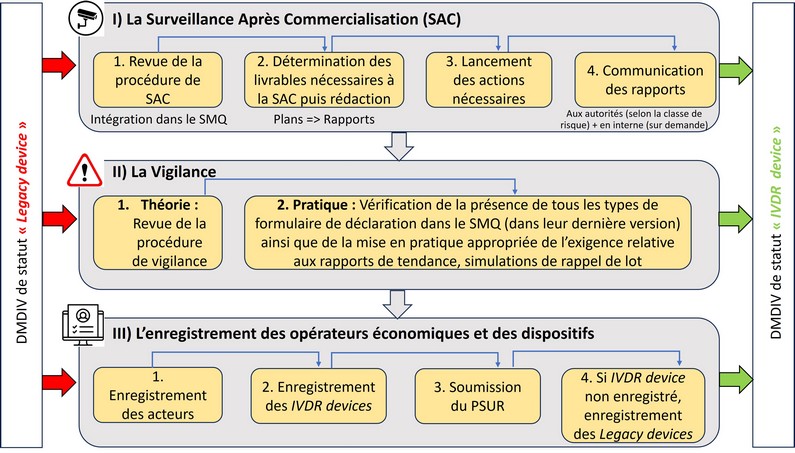
Figure 2 - Exigences du Règlement européen (UE) 20147/746 non impactées par l'amendement (UE) 2024/1860 (source UTC).
Une logique "en entonnoir"
En matière de vigilance, Genotropy a suivi une logique "en entonnoir" : du plus général, avec la revue de la procédure dédiée à la vigilance, au plus spécifique avec l’application d’exigences variables selon la classe de risque du dispositif (livrables à produire, communication…) :
Chaque année, l'Université de Technologie de Compiègne organise le Rendez-vous Biomédical de l'UTC, un événement (conférences, forum métiers et table ronde) qui favorise le dialogue entre concepteurs, chercheurs, exploitants et étudiants de la medtech. C'est notamment l'occasion de découvrir des projets réalisés par les étudiants des formations biomédicales de l’UTC.
La prochaine édition aura lieu le 24 janvier 2025 sur le thème "Grossesses à risques et soins néonatals".
- Il a été vérifié que la personne en charge de garantir la conformité des activités de vigilance est la Personne Chargée de Veiller au Respect de la Règlementation de l’entreprise, comme pour les activités de surveillance après commercialisation. L’interprétation des exigences relatives aux rapports de tendance par l’entreprise a été réfléchie dans la procédure, car il s’agit d’un processus permettant la détection des incidents non graves avant leur aggravation.
- Il a été vérifié que le SMQ comprend l’ensemble des formulaires à jour nécessaires aux déclarations aux autorités compétentes des incidents de vigilance, se produisant lors des études de performances et après la mise sur le marché, dans les délais de notification réglementaires.
EUDAMED et ses modules
Enfin, la conformité de l’entreprise aux exigences des articles 26 et 28 du Règlement (UE) 2017/746 en matière d’enregistrements à réaliser sur la base de données EUDAMED a été vérifiée. Le Règlement (UE) 2024/1860 rend obligatoire l’utilisation d’EUDAMED par module déclaré "pleinement fonctionnel", et non pas dès lors que tous les modules sont disponibles.
Ainsi, 5 des modules sur 6 prévus devraient être déclarés pleinement fonctionnels vers la mi-2025. Dans les 6 mois suivant la publication de l’avis de la Commission européenne annonçant la fonctionnalité d’un module, le fabricant devra avoir rempli ses obligations vis-à-vis de celui-ci. Pour le vérifier, Genotropy a principalement fait appel au document disponible sur le site de la Commission européenne qui pose les spécifications fonctionnelles d’EUDAMED.
Genotropy a bénéficié de l’extension de 2 ans de la fin de la période transitoire de son dispositif, en priorisant l’évaluation des performances tout en s’appliquant à la mise en œuvre de mesures déjà imposées par la réglementation.
Intégrer les recommandations de la dernière révision du guide MDCG 2021-4 (09/2024) sur l‘application des dispositions transitoires pour certifier un DMDIV de classe D est la prochaine étape de la démarche.




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





