


Actualités de la profession > Evénements
Les impacts du RDM sur la recherche clinique : deux exemples concrets

Le groupe DM de l'AFCROs effectue ici une synthèse de la 10ème Journée de la Recherche clinique qui s'est tenue en mars dernier. L'événement incluait notamment un atelier lors duquel les participants ont pu réfléchir à la mise en place d'une stratégie clinique dans deux cas concrets de DM de classe III.

Odile Capronnier

Christophe Clément
Par Odile Capronnier et Christophe Clément du groupe AFCROs-DM
Le 17 mars dernier s’est tenue la 10ème Journée de la Recherche Clinique de l’AFCROs1 au cours de laquelle un atelier animé par le groupe de travail "Dispositif Médical" portait sur l’impact de l’entrée en vigueur du Règlement Européen du Dispositif Médical (MDR – 2017/745), effective au 26 mai 2021. Si ce nouveau contexte réglementaire s’impose à tous les acteurs du secteur, les expériences des uns et des autres sont diverses et variées. Une première partie de l’atelier s’est donc attachée à partager un retour d’expérience de deux industriels du secteur, avec l’intervention de Cécile Fouret, Directrice Affaires Scientifiques chez Medtronic, et Solange Van de Morteele, Responsable Affaires Cliniques chez Teknimed.
Le MDR remplace la loi Jardé
Dans son intervention, Cécile Fouret a rappelé le contexte juridique et réglementaire applicable à la recherche clinique. Jusqu’en mai 2021, la loi Jardé et ses arrêtés étaient les textes de référence du Code la Santé Publique (CSP) pour les recherches impliquant la personne humaine. Pour les dispositifs médicaux (DM), depuis le 26 mai 2021, le texte de référence en application est le MDR et notamment le chapitre VI – Evaluation clinique et investigations cliniques. Cependant, ce qui n’est pas couvert par le MDR relève du CSP. L’impact pour les investigations cliniques soumises à compter de cette date reste limité puisque les grands principes sont identiques. Il est à noter néanmoins, entre autre :
- une nouvelle catégorisation des recherches,
- l’instauration de la notion de "procédure additionnelle invasive ou lourde" (à justifier et documenter),
- des délais et périmètres de vigilance précisés selon les articles 80 à 90 du MDR (selon le type d’investigations cliniques) en complément de l’avis aux promoteurs de l’ANSM (Agence nationale de sécurité du médicament et des produits de santé) "Partie IV Vigilance",
- l’information de la fin de l’investigation clinique selon l’article 77 du MDR.
Malgré la mise en application du MDR, il est constaté une hétérogénéité dans l’interprétation des procédures à suivre entre les pays de l’Union Européenne, par exemple la notification des cas d’Evénements Indésirables Graves ou les procédures d’autorisation par les Autorités Compétentes ou les Comités d’éthique. On peut cependant considérer que ces différences s’atténueront avec le temps, l’intégration du MDR, et la mise en œuvre prochaine de la base de données Eudamed, outil fort attendu pour la standardisation des procédures et des revues.
Un niveau d'exigence accru des organismes notifiés
De son côté, Solange Van de Morteele a présenté une synthèse des impacts de l’entrée en vigueur du MDR sur les documents constitutifs du dossier de marquage CE, et notamment sur l’évaluation clinique.
Les grands principes et les documents restent les mêmes, avec cependant quelques nouvelles parties/sections à couvrir :
- le SSCP2, dont le but est de présenter un résumé des données de sécurité et de performance à destination des ON3 et/ou des patients.
- le PSUR4, dont le but est de présenter une synthèse régulière des données de sécurité,
- le PMCF Plan5, dont la finalité est de présenter le plan de surveillance clinique du dispositif médical après sa commercialisation.
Le retour d’expérience, partagé entre les intervenants, fait état d’un niveau d’exigence réglementaire accru des ON, qui impacte les partenaires (CROs, consultants…) et les médecins prescripteurs. Par exemple, toute revendication d’utilisation d’un DM dans une indication ou une population particulière doit désormais s’appuyer sur des données cliniques.
On note également une exigence accrue de cohérence entre tous les documents constitutifs du dossier technique tels que :
- la notice d’utilisation,
- le Rapport d’évaluation clinique (CER),
- ou l’analyse de gestion des risques.
En cas d’incohérence entre les documents, notamment sur les risques résiduels, un ON peut demander une mise en conformité. De même, les objectifs envisagés dans le plan de PMCF doivent correspondre aux revendications du DM. Il s’agit donc d’aligner les données cliniques et les revendications du dossier technique afin de disposer d’une cohérence générale.
Un exercice d'application quant à la meilleure stratégie clinique à adopter
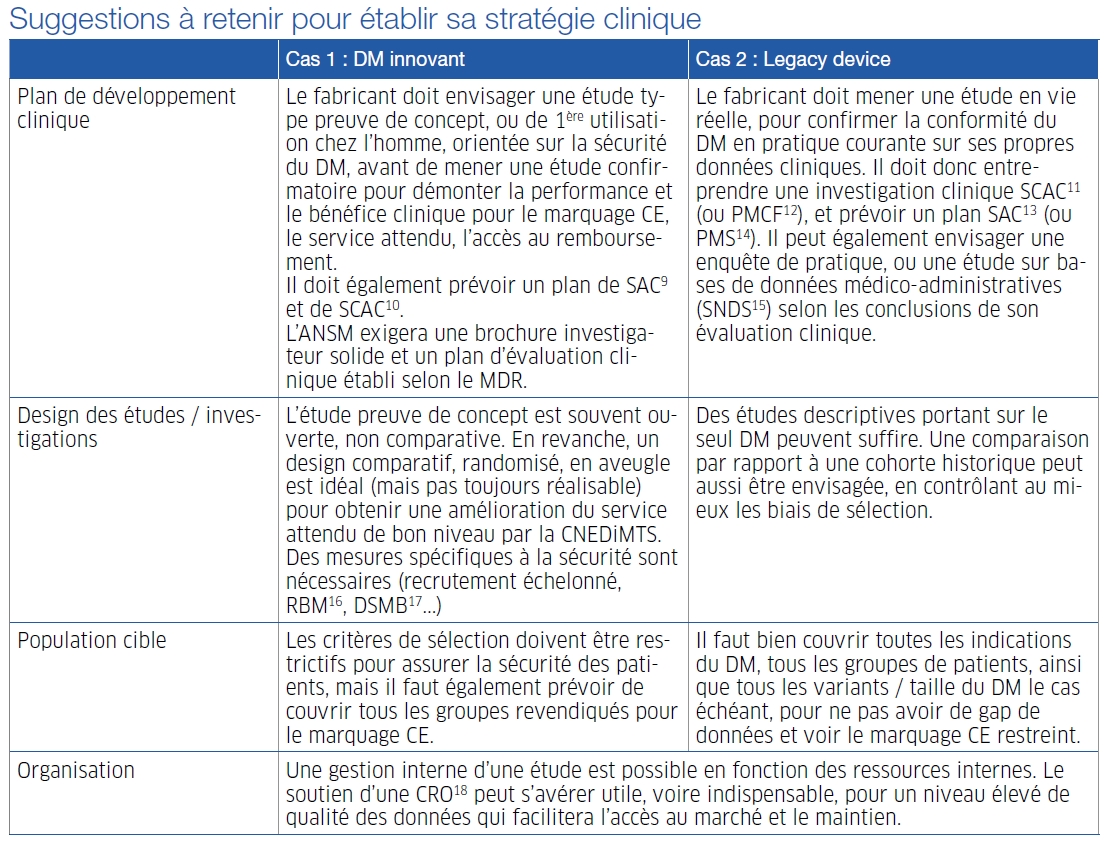
Dans la seconde partie de l’atelier, des mises en situation ont été proposées aux participants. Il s'agissait ici de se mettre dans la peau du Responsable des Affaires Cliniques devant mettre en place une stratégie clinique adaptée à son DM implantable de classe III pour répondre aux attentes des instances de contrôle :
- l’ANSM6 et le CPP7 pour l’autorisation des investigations,
- l’ON pour l’évaluation clinique et l’obtention du marquage CE,
- la CNEDiMTS8 pour le remboursement.
Le premier cas pratique était celui d'un DM innovant, développé par une start-up. Les enjeux consistaient ici à obtenir le marquage CE sous le MDR 2017/745, se conformer aux obligations de surveillance et suivi clinique post-marché, et obtenir l'inscription sur la LPPR (liste des produits et prestations remboursables) en nom de marque.
Le second cas étudié était celui d'un Legacy Device, c'est-à-dire un DM commercialisé depuis plusieurs années et ayant obtenu son marquage CE selon la Directive 93/42 sur la base de l'équivalence. L'enjeu était de maintenir son remboursement sur sa ligne générique.
Les participants à l’atelier ont réfléchi aux divers aspects de la stratégie clinique, avec l’appui des membres du groupe DM de l’AFCROs qui les orientaient à chacune des tables, avant qu’une restitution collective soit faite par les animateurs.
Les principales idées à retenir sont résumées dans le tableau ci-dessous.
[Source des illustrations : AFCROs]
1 Association Française des CROs
2 Summary of Safety and Clinical Performance
3 Organisme notifié
4 Periodic Safety Updated Report
5 Post-Marketing Clinical Follow-Up Plan
6 Agence Nationale de Sécurité du Médicament et des produits de santé
7 Comité de Protection des Personnes
8 Commission nationale d’évaluation des dispositifs médicaux
9 Surveillance après commercialisation
10 Suivi clinique après commercialisation
11 Suivi clinique après commercialisation
12 Post-market clinical follow-up
13 Surveillance après commercialisation
14 Post-market surveillance
15 Système national des données de santé
16 Risk-based monitoring
17 Data safety monitoring board
18 Clinical research organisation, ou prestataire de recherche clinique




 X (ex Twitter)
X (ex Twitter) LinkedIn
LinkedIn





